May 8 2015
A new study shows that it is possible to use an imaging technique called cryo-electron microscopy (cryo-EM) to view, in near-atomic detail, the architecture of a metabolic enzyme bound to a drug that blocks its activity. This advance provides a new path for solving molecular structures that may revolutionize drug development, noted the researchers.
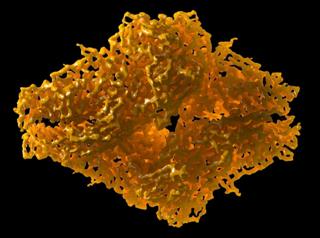 This is a 3.2 Å resolution cryo-EM structure of β-galactosidase from 2014. Credit: Veronica Falconieri, NCI
This is a 3.2 Å resolution cryo-EM structure of β-galactosidase from 2014. Credit: Veronica Falconieri, NCI
The protein imaged in this study was a small bacterial enzyme called beta-galactosidase; the drug to which it was bound is an inhibitor called phenylethyl-beta-D-thiogalactopyranoside (PETG), which fits into a pocket in the enzyme. Enzymes are typically proteins that act to catalyze biochemical reactions in the cell. Understanding what an enzyme looks like, both with and without a drug bound to it, allows scientists to design new drugs that can either block that enzyme's function (if the function is responsible for a disease), or enhance its activity (if lack of activity is causing a problem).
The study appeared online May 7, 2015, in Science Express. Sriram Subramaniam, Ph.D., of the National Cancer Institute's (NCI) Center for Cancer Research, led the research. NCI is part of the National Institutes of Health.
"This represents a new era in imaging of proteins in humans with immense implications for drug design," said NIH Director Francis S. Collins, M.D., Ph.D. "This near-atomic level of imaging provides detailed information about the keys that unlock cellular processes."
Drug development efforts often involve mapping contacts between small molecules and their binding sites on proteins. These mappings require the highest possible resolutions so that the shape of the protein chain can be traced and the hydrogen bonds between the protein and the small molecules it interacts with can be discerned.
In this study, the researchers were able to visualize beta-galactosidase at a resolution of 2.2 angstroms (or Å -- about a billionth of a meter in size), which is comparable to the level of detail that has thus far been obtained only by using X-ray crystallography. At these high resolutions, there is enough information in the structure to reliably assist drug design and development efforts.
To determine structures by cryo-EM, protein suspensions are flash-frozen at liquid nitrogen temperatures (-196°C to -210°C , or -320°F to -346°F) so the water around the protein molecules stays liquid-like. The suspensions are then imaged with electrons to obtain molecular images that are averaged together to discern a three-dimensional (3D) protein structure.
"The fact that cryo-EM technology allows us to image a relatively small protein at high resolution in a near-native environment, and knowing that the structure hasn't been changed by crystallization, that's a game-changer," said Dr. Subramaniam.
In the study, using about 40,000 molecular images, the researchers were able to compute a 2.2 Å resolution map of the structure of beta-galactosidase bound to PETG. This map not only allowed the researchers to determine the positioning of PETG in the binding pocket but also enabled them to pick out individual ions and water molecules within the structure and to visualize in great detail the arrangement of the amino acids that make up the protein.
Dr. Subramaniam and colleagues have recently used cryo-EM to understand the functioning of a variety of medically important molecular machines, such as the envelope glycoproteins on HIV and glutamate receptors found in brain cells. Their new finding, however, represents the highest resolution that they or others have achieved to date for a structure determined by cryo-EM.
"Cryo-EM is positioned to become an even more useful tool in structural biology and cancer drug development," said Douglas Lowy, M.D., acting director, NCI. "Even for proteins that are not amenable to crystallization, it could enable determination of their 3D structures at high resolution."