Oct 24 2008
Every copy of the human genome generally consists of the same set of genes, but considerable variability exists between individuals at the sequence level. Variations can include single nucleotide polymorphisms (SNPs), in which one DNA base is substituted for another; or copy-number variations (CNVs), in which entire blocks of gene sequence are deleted or duplicated. Individual variations can alter the function or activity of affected genes, and thus have important health implications.
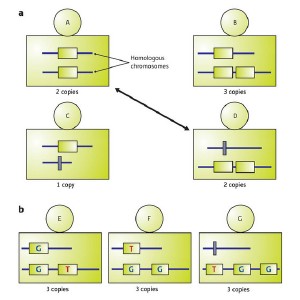 Copy-number variation (CNV) can occur in ambiguous patterns. (a) Individuals in a population may have different copy numbers on homologous chromosomes at CNV loci. For example, here individual A and D have two copies, although the patterns are different: A has one copy on each chromosome, whereas D has two on one chromosome and zero on the other. (b) Individuals may also have CNVs that contain SNPs. For example, individuals E, F, and G each have three copies, but the patterns can be distinguished by the numbers of copies on each chromosome and variations defined by SNPs.
Copy-number variation (CNV) can occur in ambiguous patterns. (a) Individuals in a population may have different copy numbers on homologous chromosomes at CNV loci. For example, here individual A and D have two copies, although the patterns are different: A has one copy on each chromosome, whereas D has two on one chromosome and zero on the other. (b) Individuals may also have CNVs that contain SNPs. For example, individuals E, F, and G each have three copies, but the patterns can be distinguished by the numbers of copies on each chromosome and variations defined by SNPs.
Also important, however, is an individual's 'haplotype'-the set of variations that co-occur on a given chromosome. Human chromosomes typically come in pairs, and the combined data from two haplotypes is referred to as a 'diplotype'; in some cases, the impact of a specific diplotype can be just as important for predicting drug response or disease susceptibility as the variations it contains.
Tools for high-throughput SNP haplotype analysis are already part of the clinical diagnostic arsenal, but equivalent methods are not as well developed for CNVs. "CNVs are getting more widely recognized as useful indicators," says Tatsuhiko Tsunoda of the RIKEN Center for Genomic Medicine in Yokohama (formerly the SNP Research Center), "but they are still challenging to handle."
Since CNV patterns can be highly complex, effective high-throughput CNV haplotype analysis will require sophisticated mathematical strategies. To this end, Tsunoda's group developed a computational tool called CNVphaser, which applies an algorithm to CNV datasets to infer the most probable individual haplotypes*. Their system can perform calculations based on information about each individual's combined copy number for both chromosomes or, for CNVs that also contain SNP variants, based on the frequency of occurrence of individual nucleotides within this set of copies.
CNVphaser accurately estimated haplotype frequencies in initial tests using simulated genomic datasets, and the team noted that the algorithm was especially effective for analyzing variant regions in which a small number of specific haplotypes occur with much greater frequency than other potential haplotypes.
The algorithm also performed well in a real-world test, in analyses of gene variants from two distinct human populations-Nigerian Yoruba and individuals of European ancestry in Utah. "CNV patterns showed quite different frequencies-up to dozens of percentage points-between the two populations," says Tsunoda. "This shows the algorithm's big power for identifying differences in copy patterns between two groups." Based on this initial success, Tsunoda is optimistic that his method will also prove valuable in the clinic, and is now exploring its effectiveness for performing genetic association studies with patient samples.
- Kato, M., Nakamura, Y. & Tsunoda, T. An algorithm for inferring complex haplotypes in a region of copy-number variation. The American Journal of Human Genetics 83, 157-169 (2008).