Mar 3 2011
Indiana University scientists have solved a perplexing mystery regarding one of the body's main energy storage molecules, in the process shedding light on a possible route to treatment of a rare but deadly disease in teenagers.
The disease occurs when a genetic mutation causes excessive amounts of phosphate to build up in glycogen. Glycogen is a chain-like molecule the body uses to temporarily store glucose when it's not needed to provide energy for cellular activities. The excess phosphate causes unnatural glycogen structures to appear in the body, including the brain, resulting in progressive neurological problems.
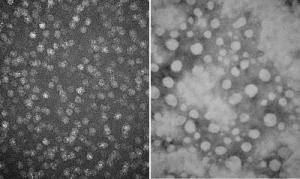 Electron microscopy images demonstrate how glycogen molecules (left) become structurally abnormal (right) when excessive levels of phosphate are attached due to a mutation in the lafora gene.
Electron microscopy images demonstrate how glycogen molecules (left) become structurally abnormal (right) when excessive levels of phosphate are attached due to a mutation in the lafora gene.
In a paper in the March 2, 2011 issue of the journal Cell Metabolism, a research team led by Peter J. Roach, Ph.D., professor of biochemistry and molecular biology at the IU School of Medicine, has identified where the extra phosphate comes from, and how it is chemically linked to the glycogen molecules. The findings, Dr. Roach said, suggest a possibility for treating the disease – stopping the body from producing glycogen.
Lafora disease, named after the Spanish physician who identified it, strikes early in the teen years with epileptic seizures and then other neurological symptoms that grow progressively worse. It is always fatal, usually within 10 years.
"It's a very harrowing disease and there's no treatment right now. It's thankfully rare, but for the families affected it's tragic," said Dr. Roach, Distinguished Professor and a Chancellor's Professor, Indiana University-Purdue University Indianapolis.
The disease is caused by a mutation to one of two genes, one leading to a defective version of a protein called laforin. In previous research, Dr. Roach and his collaborators showed that laforin's role is to remove phosphate residues from glycogen. When the body cannot produce laforin, the phosphate levels in glycogen build up, resulting in Lafora disease.
What had puzzled scientists was the source of the phosphate. The answer, Dr. Roach and his colleagues report in Cell Metabolism, is found in the enzyme that cells normally use to build up the glycogen molecules. When there are extra glucose molecules in the body, such as after a meal, the enzyme, like a tiny molecular machine, hooks the glucose molecules onto the glycogen chain. In the process, phosphates that are attached to the glucose are discarded.
But once every 10,000 cycles or so, a phosphate molecule remains attached when the enzyme hooks the glucose molecule onto the glycogen chain. The job of laforin is to correct for those errors. Without the laforin, phosphates build up and the deadly disease is the result.
Dr. Roach and his colleagues note that animal experiments suggest that a treatment to counteract the laforin mutation could be compounds that would block the production of glycogen. Although a lack of glycogen could have side effects – such as greater propensity to develop diabetes – they might be acceptable in the face of such a deadly alternative, Dr. Roach said. However, such an approach is only a theoretical possibility at his point, he said.
Source: http://www.medicine.iu.edu/