Hydrogen energy has the potential to meet the United Nations net zero emissions target; however, its industrial application has been stalled by the control and storage difficulties.
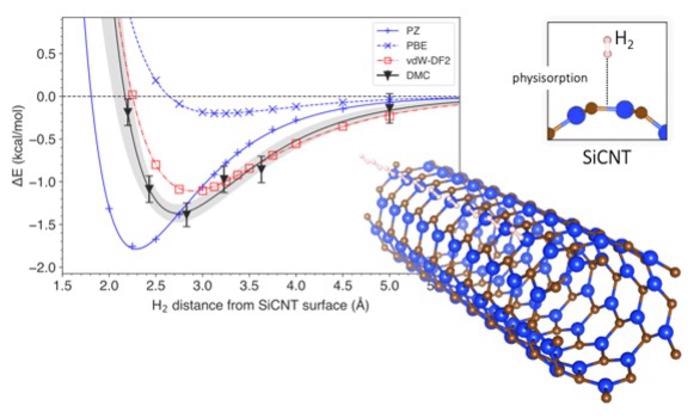 The graph shows the variation of system energy with the distance of a hydrogen molecule from the surface of a silicon carbide nanotube (bottom right). The depth of the curve signifies the energy required to extract hydrogen from storage. A comparison of prediction methods is presented, with DMC being the most accurate and vdW-DF2 being its closest match. Image Credit: Kenta Hongo from JAIST.
The graph shows the variation of system energy with the distance of a hydrogen molecule from the surface of a silicon carbide nanotube (bottom right). The depth of the curve signifies the energy required to extract hydrogen from storage. A comparison of prediction methods is presented, with DMC being the most accurate and vdW-DF2 being its closest match. Image Credit: Kenta Hongo from JAIST.
Hydrogen turns into a gas at a very low temperature (−252 °C), making its storage at ambient temperature difficult. The interaction between hydrogen and its storage material is too weak to persist at room temperature. This makes the design of storage materials vital to accomplishing the goal of transforming hydrogen energy into everyday use.
Computational materials design has the solution. Time and effort can be reduced during the creation of hydrogen technology by engineering a material on a computer and mimicking its capability for hydrogen storage. Yet, the predictions become very restricted in their application unless they are accurate and can be made at a reasonable computational cost.
In a new study reported in ACS Omega, researchers developed a computationally costly, but very accurate, novel process for predicting hydrogen storage.
Improving prediction reliability for simulations can help accelerate the development of materials for hydrogen fuel storage and lead to a more energy efficient society.
Dr. Kenta Hongo, Study Lead, Japan Advanced Institute of Science and Technology
The van der Waals force is one of the major forces of attraction between objects, which describes the interaction between molecules or atoms according to the distance between them. Since the Van der Waals force is the result of quite complex quantum procedures, conventional treatments could not define it properly, and hence the simulations thus far are at the level of approximate estimations of it.
But, was it correct to do so when mimicking the storage of hydrogen? This was the key concern of Dr. Hongo and his team.
To find a solution to this question, they analyzed silicon-carbide nanotubes, one of the most favorable materials for hydrogen storage. Using a computational method called diffusion Monte Carlo (DMC), they designed a model that took into account van der Waals forces when mimicking hydrogen storage in silicon-carbide nanotubes.
A majority of the traditional models look at the interactions between silicon-carbide nanotubes and hydrogen as a whole, but the DMC technique utilizes the supercomputer’s power to rebuild the interaction mechanism realistically by following the configuration of individual electrons. This renders the DMC model the most accurate approach of prediction thus far.
Using the DMC model, the team was also able to estimate how much energy would be needed to remove hydrogen from its storage and at what distance the hydrogen was expected to be from the surface of the silicon-carbide nanotube. They then compared the outcomes from their modeling to those acquired through conventional prediction approaches.
Conventional prediction approaches are typically based on computational methods known as the density functional theory (DFT). DFT uses functionals, which are model descriptions of quantum interactions that define the spatial differences of electron density to establish the properties of complicated systems.
While there have been a number of DFT-based studies on hydrogen storage on silicon-carbide nanotubes, none of them have integrated van der Waals forces into their estimations. Van der Waals-corrected DFT functionals have, however, been employed in the estimation of other materials.
Dr. Hongo and the other researchers replicated hydrogen storage using a broad range of DFT functionals, some with van der Waals corrections and some without. They discovered that the DFT functionals without van der Waals corrections wrongly estimated the energy necessary for the storage of hydrogen by 4%–14%.
Conversely, van der Waals-corrected DFT functionals generated results that were fairly similar to those of DMC. Furthermore, they learned that the influence of the van der Waals force on the storage energy was around 9%–29%, which is hardly irrelevant.
These discoveries, Dr. Hongo believes, can pave the path for further modernization in hydrogen storage simulation technology.
Although the DMC method is computationally expensive, it can be used to clarify the peculiarities (tendencies of prediction error) of each prediction method. This will help us understand which prediction to trust, and also how to modify prediction methods to make them more useful.
Dr. Kenta Hongo, Study Lead, Japan Advanced Institute of Science and Technology
With robust computational estimations like this to accelerate its development, one can soon see hydrogen technology becoming a daily reality.
Journal Reference:
Prayogo, G. I., et al. (2021) Importance of van der Waals interactions in hydrogen adsorption on a silicon-carbide nanotube revisited with vdW-DFT and quantum Monte Carlo. ACS Omega. doi.org/10.1021/acsomega.1c03318.