Feb 5 2009
A RIKEN-led team of molecular biologists has determined the specific bonding leading to the formation of a protein complex involved in distributing pigment throughout the skin. Disruption of this membrane transport complex leads to the rare, lethal Griscelli syndrome for which there is no effective treatment. Patients show symptoms of albinism, suffer immunodeficiency, and typically die in early childhood. The work may stimulate the development of therapeutic drugs for the condition.
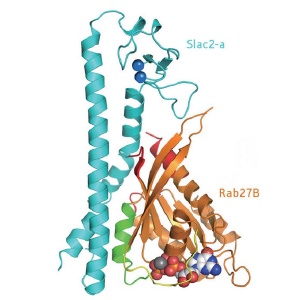 Diagram of the structure of the Rab27B/Slac2-a complex.
Diagram of the structure of the Rab27B/Slac2-a complex.
Members of the Rab protein family—of which there are more than 60 in humans—are thought to be essential to membrane trafficking, an important form of communication and distribution within cells. Rab proteins are typically bound to membranes either in an inactive guanine diphosphate form or an active triphosphate form which works through specific effector molecules to promote membrane trafficking.
The pigment melanin, which protects against radiation damage, is made and distributed within vesicles called melanosomes in skin color cells known as melanocytes. Rab27, which comes in two forms A and B, binds into a complex with the effector protein Slac2-a and myosin Va to transfer melanosomes onto actin filaments. The complex then transports the melanosomes along the filaments to where Rab27 uses another effector molecule to anchor them to the outer membrane of the cell.
Researchers from the RIKEN Systems and Structural Biology Center in Yokohama together with colleagues from Tohoku University were able to crystallize the Rab27B/Slac2-a complex and solve its structure using x-ray diffraction. As active Rab27 proteins are notoriously difficult to crystallize, this was the first mammalian complex where the binding of such a protein with its effector molecule could be thoroughly investigated. The results were published recently in the journal Structure*.
The researchers found three contact regions between Rab27B and Slac2-a, of which only one was involved in specific recognition. Mutations affecting any of the several specific intermolecular hydrogen bonds in this region were fundamentally disruptive, and some of them led to Griscelli’s syndrome. The group was able to verify the structure by taking another Rab protein, Rab3A, and engineering it to bind Slac2-a. The Rab3A amino acid sequence had to be altered by only four amino acid residues in the critical binding area to form the complex with Slac2-a.
“We are hoping that pharmaceutical companies will be able to use our structure as a basis for drugs which can be used to treat conditions like Griscelli’s syndrome,” says first author Mutsuko Kukimoto-Niino.
*Kukimoto-Niino, M., Sakamoto, A., Kanno, E., Hanawa-Suetsugu, K., Terada, T., Shirouzu, M., Fukuda, M. & Yokoyama, S. Structural basis for the exclusive specificity of Slac2-a/melanophilin for the Rab27 GTPases. Structure 16, 1478–1490 (2008).